|
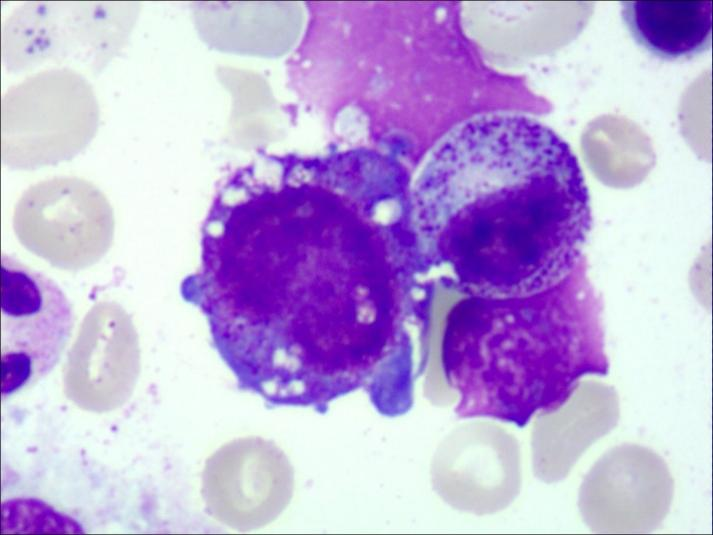
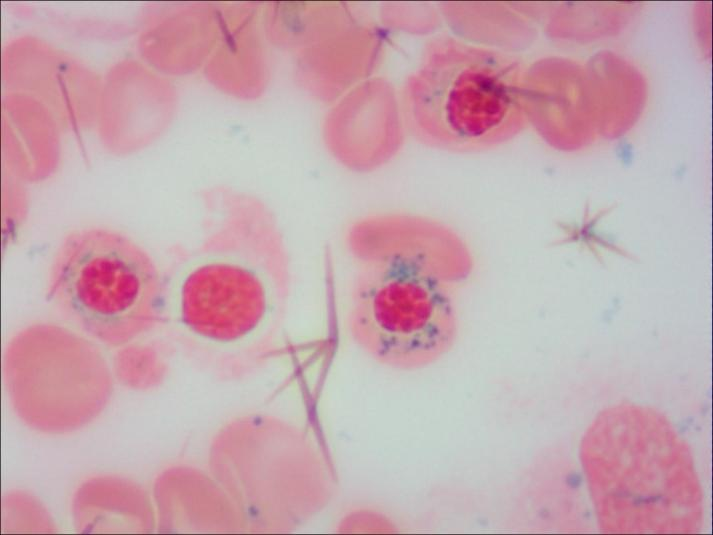
近日,中南大学湘雅二医院儿童医学中心血液肿瘤专科收治了一名以“反复发热、难治性贫血、生长发育落后”为临床表现的2岁女童,住院后结合骨髓细胞学与线粒体基因检测诊断为罕见病――“皮尔逊综合征”。经过治疗,患儿目前整体情况稳定,但仍有不同程度贫血,需长期于该院儿科血液肿瘤专科门诊随访并定期行红细胞输注及代谢调节治疗。 今年2月28日是第十八个国际罕见病日,今年的主题是“不止罕见(More than you can imagine)”,旨在进一步提高大众对罕见病的关注和认知,推进罕见病科普宣教,促进罕见遗传病的筛查、诊断及预防,提高生存质量。在众多罕见病中,皮尔逊综合征全球报告病例不超过150例,因其复杂性和对儿童的致命威胁,亟需更多认知与行动。 皮尔逊综合征(Pearson Syndrome,PS),是一种由线粒体DNA缺失引发的原发性线粒体病,发病率约为百万分之一。其发病机制涉及遗传缺陷、代谢紊乱及组织异质性等多方面因素,患儿发育过程中这种线粒体DNA缺失在各系统组织中的累积不同,从而引起不同的临床表现。它像一道隐形的裂缝,悄然侵袭患儿的血液系统、胰腺及多个器官,多数患儿在出生后6个月内发病,被称为“生命最初的生存挑战”。 据悉,患儿初期常表现为不同程度的贫血,大多面色苍白;伴或不伴有白细胞和血小板低下,感染及出血风险极高; 部分患儿伴有胰腺外分泌功能不全,主要表现为腹泻、营养不良,需依赖消化酶替代治疗;主要表现为乳酸中毒,可导致多器官功能的障碍;患儿的身高、体重多低于同年龄段其他儿童。此外,肝脏、肾脏、神经系统可能出现进行性的损伤,部分患儿后期甚至可能发展为更严重的Kearns-Sayre综合征(眼肌麻痹、视网膜病变)。 由于症状复杂,皮尔逊综合征常被误诊为普通贫血或感染,确诊依赖骨髓细胞学检查和线粒体DNA分析。 目前,确诊方法通过基因检测,即通过血液或骨髓样本检测线粒体DNA大片段缺失;骨髓活检,即可以观察到特征性的红系前体细胞、粒系前体细胞的胞质“空泡化”以及环形铁粒幼细胞。
主要采用输血和铁螯合剂,缓解贫血,预防铁过载; 胰酶和代谢支持,改善消化吸收,补充维生素及辅酶以改善机体代谢;对症管理代谢问题,如纠正酸中毒。 近年来,科研领域迎来突破,如基因疗法,针对线粒体DNA缺陷的修复技术进入临床试验阶段;线粒体移植,首个线粒体增强疗法(MAT)临床试验启动(ClinicalTrials.gov 标识符:NCT03384420),给患者和家庭带来了新的希望;科研助力全球皮尔逊综合征相关论文较少,中国学者正领跑基因治疗。(胡素云 黄尔佳) |
热门关键词: